Members from the GRC attended the 10th International Zebrafish Genetics and
Development meeting in Madison, Wisconsin, to gain insight into trends of
current research and provide information to the community. New approaches for
the identification and targeting of mutations (e.g. Whole Genome Sequencing and
the use of Transcription Activator-Like Effector Nucleases (TALENs)) were a
recurring theme throughout the meeting, highlighting the importance of improved
reference assemblies with accurate sequence. Whole genome sequencing is being
used in forward genetic screens for the identification of mutations, whereas
TALENs are being used to generate targeted gene mutations in zebrafish. A high
quality reference genome assembly is of utmost importance to the success of
both of these technologies to allow accurate mapping for experimental design (oligonucleotide
design for TALENs) and subsequent data analysis (mapping whole genome sequence
data to the reference for mutation identification). Discussions held at the
meeting indicated that the zebrafish research community would welcome a new
assembly.

Fig.
1:
A PGPviewer screenshot from the Zv9 and GRCz9b assemblies, showing localisation
of a previously unlocalised scaffold. In Zv9, FP236471 was not localised to a
chromosome and remained on a single scaffold (Scaffold
Zv9_scaffold3539), whereas FP003601 was located to chromosome 4. The red block
at the end of FP236471, in Zv9, indicates sequence similarity between the
two clones. In GRCz9b, an overlap has been made, adjoining the two contigs,
with a perfect sequence alignment, indicated by the green clone overlap block
at the bottom of the screenshot. The feature tracks have been kept to a minimum
for illustration purposes.

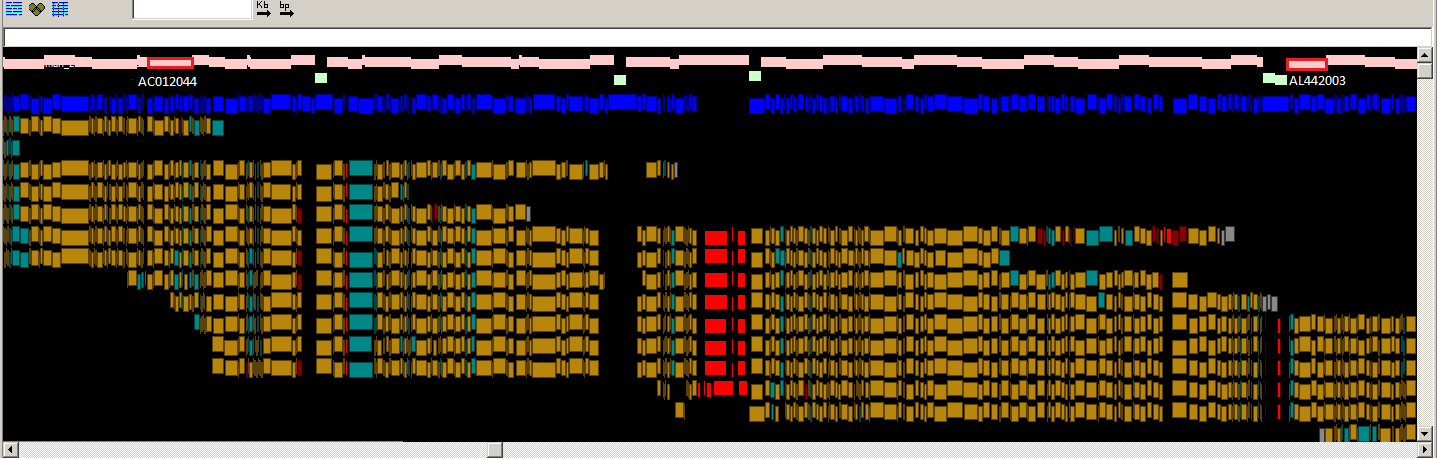
Fig.
2: A PGPviewer screenshot from the Zv9 and GRCz9b assemblies,
showing the correction of an over-expanded region in Zv9. In Zv9, CU571256 and
BX927241 reside next to each other with no overlap. However, the red blocks
show a potential alignment between them and the end sequence alignments also
indicate an over-expansion. In GRCz9b, the clones overlap with a highly
variable alignment due to the haplotypic nature of the two libraries used, as
highlighted by the red clone overlap block at the bottom of the screenshot. The
feature tracks have been kept to a minimum for illustration purposes.
Approaches underway, to complement existing
genome curation, include increasing the coverage of SATMAP, the high-density
meiotic map used to allocate all genomic contigs, along with the sequencing of
more than 1000 genomic clones to fill gaps and cover those genes still missing.
To ensure effective utilization of the new clone sequence, GRCz10 will contain
unfinished sequence with HTGS phase 2 (ordered and oriented contigs), unlike
its predecessor. The GRC are planning to release GRCz10 in 2013.